이상수의 Health Policy Insight
[Health Policy Insight 504호]

메드트로닉코리아 부사장
US FDA는 매년 약 3,000개의 새로운 의료기기를 승인한다. FDA는 새로운 의료기기에 대한 경쟁적 우선순위, 즉 신기술에 대한 환자의 적시 접근성과 편익 및 위험 간의 과학적 확실성에서 균형을 맞춰야 한다. FDA가 올바른 균형을 잡을 수 있는지에 대한 논란이 계속되고 있다. 산업계와 일부 환자단체는 FDA가 과도하게 부담스러운 근거 요건을 부과하고 시판 전 심사를 장기화해 기술 혁신과 환자 접근성을 저해한다고 주장해 왔다. 이들은 과도하게 엄격한 규제의 결과로 의료기기 개발에 대한 투자가 부적절하고 미국 시장 진입 전에 해외에서 새로운 의료기기가 빈번하게 출시되는 것을 꼽는다. 이에 반해 국립 의학원(National Academy of Medicine National Academy of Medicine)을 비롯한 다른 단체들은 FDA 요건이 의료기기의 안전성 및 효과성을 보장하기에 충분하지 않다는 우려를 표명했다.
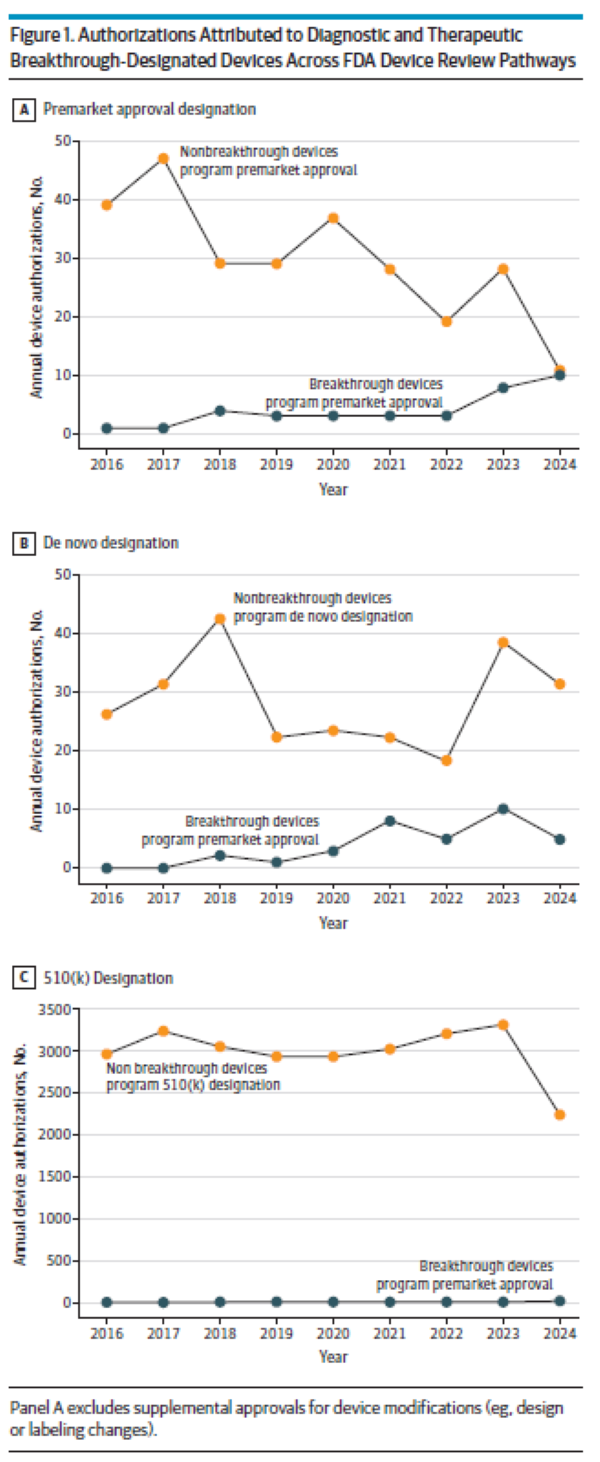
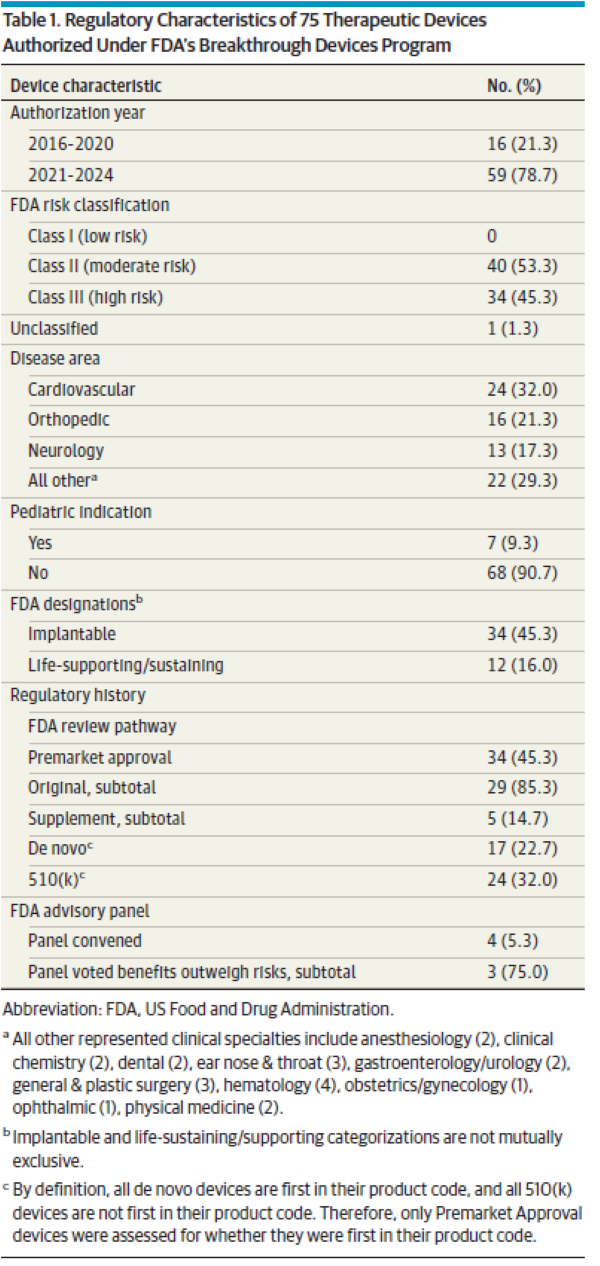
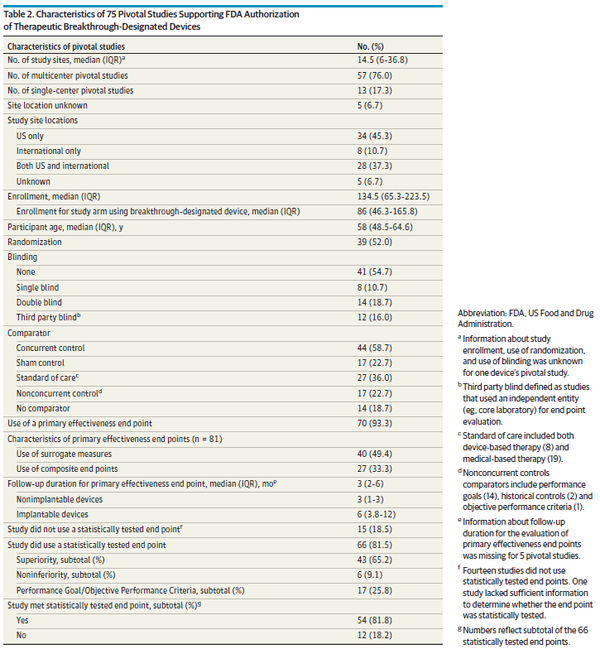
의료기기 혁신과 환자 접근성을 촉진하기 위해 의회는 2016년 21세기 치유법(21st Century Cures Act)에 따라 혁신 의료기기 프로그램(Breakthrough Devices Program, BDP)을 제정했다. 제조업체는 시판 전 개발 중 언제든지 혁신 의료기기 지정을 요청할 수 있으며 FDA는 이를 승인할 수 있다. 모든 FDA 위험도 분류(낮음[1등급], 보통[2등급] 또는 높음[3등급])의 의료기기는 2가지 추가 기준을 충족하는 경우 혁신 의료기기 지정을 받을 수 있다. 첫째, 의료기기가 생명을 위협하거나 비가역적으로 쇠약하게 만드는 질환에 대해 보다 효과적인 치료 또는 진단을 제공해야 한다. 둘째, 해당 의료기기는 △획기적인 기술이어야 하고 △승인된 대안이 없으며 △기존 기술 대비 상당한 이점을 제공하거나 △환자에게 최선의 이익이 돼야 한다. BDP에 참여하면 자격을 갖춘 제조업체는 시판 전 개발을 촉진하기 위한 다양한 규제 혜택(FDA 담당자와의 협의 확대, 유연한 시판 전 및 시판 후 임상연구 설계 사용, FDA 우선 검토(prioritized review))을 받을 수 있다. BDP는 출범 이후 크게 성장해 수많은 의료기기가 지정 및 후속 FDA 승인을 받았다. 초기 보고서에서는 혁신 지정 의료기기가 진정한 혁신 기술인지에 대한 우려를 확인했지만, BDP가 시기적절하게 혁신적인 의료기기에 접근할 수 있는지 결정하려면 현대적이고 종합적인 평가가 필요하다. FDA가 승인한 모든 혁신 지정 의료기기를 검토한 결과, 대부분의 의료기기가 최초이며 의회에서 정한 목표기간 내에 FDA 심사를 받은 것으로 나타났다. 그러나 1/10개의 의료기기는 임상시험 없이 임상진료에서 사용할 수 있게 됐다. 또한 많은 혁신 지정 의료기기를 뒷받침하는 근거에는 상당한 한계가 있었는데, 1/3개 의료기기는 대리 측정치(surrogate measures)를 1차 평가변수(primary endpoint)로 사용했고, 1/8개는 비교 효과성(comparative effectiveness) 시험이 부족했으며, 1/7개는 1차 유효성 평가변수를 충족하지 못했다. 이런 결과는 BDP가 기존 대안 대비 상당한 이점을 제공하는 의료기기 개발을 촉진하라는 의회의 명령을 제대로 이행하고 있는지 의문을 제기한다.
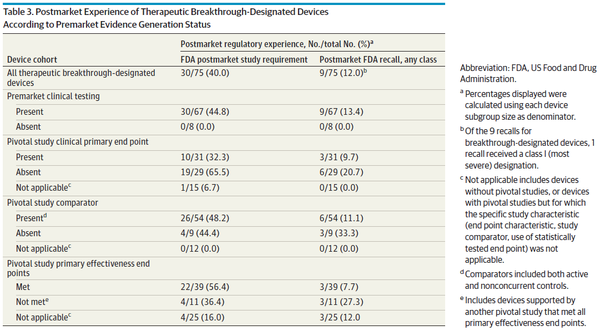
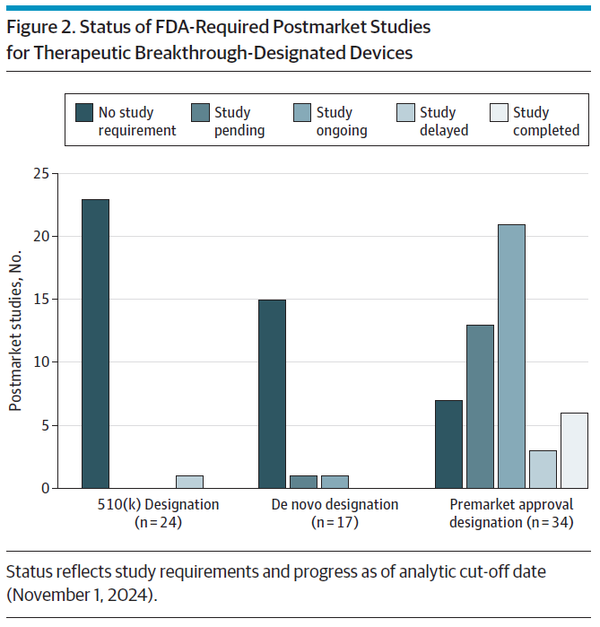
연구 결과에 따르면, BDP는 과거 FDA 프로그램보다 제조업체의 참여를 촉진하고 의료기기 출시를 더 신속하게 촉진한 것으로 나타났다. 1991~2015년에 FDA는 신속 심사(expedited review)를 통해 총 178개 의료기기(연간 평균 7.0개 의료기기)를 승인했다. 반면, BDP에서는 이미 127개 의료기기(연간 평균 14.5개 의료기기)를 승인했으며, 900개 이상의 혁신 지정 의료기기가 아직 시판 전 개발 단계에 있다. 고위험 의료기기의 경우, BDP에 따라 FDA 검토 기간은 이전의 우선 검토 프로그램(Priority Review Program, 평균 1.8년)보다 훨씬 더 짧았다(평균 0.7년). 또한 대부분의 혁신 지정 의료기기에 대한 FDA 검토기간은 의료기기 사용자 수수료(Medical Device User Fee Authorization, MDUFA) 성과목표(performance goal)를 충족했지만, 이런 목표는 주로 혁신 지정이 아닌(즉, 전반적으로 기술 또는 과학적으로 덜 새로운) 의료기기에 대한 기대치를 기반으로 한 것이었다. 일부에서는 제조업체 참여 증가와 검토기간 단축을 더 많은 의료기기에 대한 조기 접근이 환자 결과를 개선할 것이라는 가정 하에 인용할 수 있다. 그러나 이번 연구 결과는 혁신 지정 의료기기에 대한 규제 선택성, 속도, 엄격성 간의 상충관계를 잘 보여준다. 예를 들어, 기존 의료기기와의 실체적 동등성(substantial equivalence) 및 비임상 데이터를 기반으로 510(k) 경로를 통해 승인된 의료기기가 궁극적으로 환자 결과를 개선할지는 여전히 불분명하다. 또한, 많은 확증 임상연구(pivotal study)가 대리 평가변수에 의존했지만 이런 측정치가 임상결과 개선을 항상 정확하게 추정하는 것은 아니다. FDA는 시판 후 데이터 수집을 통해 허가 당시의 잔존 불확실성을 해결하고자 한다고 밝혔다. 그럼에도 불구하고 시판 전 임상시험 없이 허가된 모든 의료기기와 확증 임상연구가 1차 평가변수에 실패한 대부분의 의료기기를 포함해 혁신 지정 의료기기의 60%에 대해 시판 후 연구를 요구하지 않은 것으로 나타났다. FDA가 시판 후 연구를 요구한 경우에도, 이런 연구는 주로 대리 평가변수를 사용했기 때문에 시판 후 데이터가 혁신 지정 의료기기의 안전성 및 효과성을 검증하기에 충분한지 의문이 제기됐다. 이용 가능하고 적절한 확증 임상연구가 부족하면 치료 결정을 내리려는 환자와 의사에게 불확실성이 지속될 수 있다.
의회와 FDA는 BDP에 대한 몇 가지 개혁을 고려할 수 있다. 첫째, FDA는 이전에 제조업체 요청의 71%를 승인한 것으로 보고된 바 있는 혁신 지정을 보다 선별적으로 부여하는 방안을 고려할 수 있다. FDA는 혁신 지정 자격을 얻기 위해 의료기기가 효과성에 대한 통계적 검증에 따라 1차 평가변수를 사용해 시판 전 임상시험을 수행할 계획이 있어야 한다고 명시할 수 있다. 둘째, FDA는 허가 시 혁신 지정의 근거를 공개해 혁신 지정이 반드시 의료기기의 안전성 및 효과성에 대한 강건한 근거를 의미함으로써 환자와 의사의 부주의한 오해를 해소할 수 있다. 셋째, FDA는 기존 권한을 행사해 특히 확증 임상연구가 1차 평가변수를 충족하지 못하거나 대리 측정에 의존하는 경우 더 많은 의료기기에 대해 임상결과에 초점을 맞춘 시판 후 연구를 요구할 수 있다. 넷째, 의회는 FDA에 허가 시 시판 후 연구 개시를 요구하고 시판 후 연구에서 임상적 혜택을 확인하지 못한 경우 혁신 지정을 취소할 수 있는 권한을 부여할 수 있다. FDA는 이미 신속 심사 프로그램(expedited review program)에 따라 승인된 의약품에 대해 이런 권한을 가지고 있으며, 현재까지 약 15%의 혁신 지정 의약품(breakthrough-designated drug)에 대해 지정을 철회했다.
|
시사점 ㆍ의료기기 혁신과 환자 접근성을 촉진하기 위해 의회는 2016년 21세기 치유법(21st Century Cures Act)에 따라 혁신 의료기기 프로그램(Breakthrough Devices Program, BDP)을 제정함 |
* 본 컬럼은 의료기기를 비롯한 헬스케어 분야의 국내외 학회지에 발표된 논문 및 연구보고서 등을 살펴봄으로써 우리나라 의료기기 관련 보건의료정책 마련에 통찰력을 제공하기 위한 목적으로 매주 발표됨
출처 : FDA Authorization of Therapeutic Devices Under the Breakthrough Devices Program
Kadakia KT, et al. JAMA Intern Med. doi:10.1001/jamainternmed.2025.2235
https://jamanetwork.com/journals/jamainternalmedicine/article-abstract/2835682?utm_campaign=articlePDF&utm_medium=articlePDFlink&utm_source=articlePDF&utm_content=jamainternmed.2025.2235