이상수의 health policy insight
● [Health Policy Insight 291회]
US FDA의 '혁신적 의료기기 프로그램(Breakthrough Devices Program)' 초기 경험

Medtronic North Asia
(Korea and Japan)
대외협력부 전무
미국 의회는 심각한 질병의 진단과 치료를 위해 혁신적 의료기기에 대한 환자 접근성을 촉진하기 위해 2016년 12월 US FDA의 ‘획기적 의료기기 프로그램(Breakthrough Devices Program, BDP)’을 승인했다. 2019년 8월, 미국 보험청(Centers for Medicare and Medicaid Services, CMS)은 임상 채택을 촉진하기 위해 획기적 의료기기에 대한 병원 보험급여(hospital reimbursement)를 증가시키는 변경사항을 마무리했다. 프로그램이 시작된 이래, BDP는 빠르게 증가하는 속도로 성장했다: 2020년 1월 1일 현재 US FDA는 222개 의료기기에 획기성 지정(breakthrough designation)을 부여했으며 이는 1년전 대비 거의 200% 프로그램 성장을 보여준다. 의료기기 개발 및 검토기간을 단축하기 위해, US FDA는 보완적인 시판 후 데이터(post-market data)를 수집할 목적으로 덜 엄격한 시판전 근거(premarket evidence)를 기반으로 획기적 의료기기를 승인했다. 예를 들어, US FDA는 유효성 데이터를 뒷받침하지 않거나 상당한 안전성 위험이 있는 획기적 의료기기를 승인했다. 그럼에도 불구하고, 획기적 의료기기는 US FDA 승인에 앞서 수년 동안 유럽에서 상용화된다.
획기적 의료기기에 대한 규제를 강화하기 위해 US FDA는 규제기관, 특히 유럽연합의 규제기관과 협력하여 실세계 시판 후 경험을 활용해야 한다. 또한, US FDA와 CMS는 획기적 의료기기에 대한 임상연구 요건을 조정하여 메디케어 보험급여 의사결정을 촉진하고 임상적 의사결정을 뒷받침해야 한다. 이러한 조치는 BDP가 미충족 임상적 니즈를 가진 환자에게 안전하고 효과적인 의료기기를 제공하는데 도움이 될 수 있다.
BDP 프로그램과 기원(BDP program and its provenance)
고위험 의료기기(high-risk devices)의 경우, 시판전 평가의 주요 기초가 되는 연구인 중추적 임상시험(pivotal clinical trials) 시작부터 US FDA 승인까지 소요되는 기간은 일반적으로 5년을 초과한다. 의료기기 제조업체와 환자 옹호단체(patient advocacy groups)는 고위험 의료기기에 대한 US FDA 검토 프로세스가 지나치게 번거롭고 다른 국가에서 이용 가능한 기술을 포함하여 잠재적으로 생명을 구할 수 있는 의료기기에 대한 접근을 방해한다는 우려를 표명했다. 이러한 요인으로 인해 US FDA는 ‘21세기 치유법(21st Century Cures Act)’에 따라 BDP를 도입하고 CMS가 이러한 프로그램에 따라 승인된 의료기기의 보험급여를 개선하도록 했다. BDP는 의료기기 규제검토를 촉진하기 위한 기존의 2가지 특별경로(special pathways)인 ‘우선검토 프로그램(Priority Review Program, PRP)’과 ‘신속 접근성 경로 프로그램(Expedited Access Pathway Program, EAP)’을 대체한다. PRP는 US FDA 시판 전 검토 대기열에 잠재적으로 혁신적인 의료기기를 우선 배치하기 위해 2013년에 설립되었다. 그러나 이것은 새로운 기술과 과학 개념을 규제하는 것과 관련된 문제로 인해 검토를 촉진하기에는 때때로 불충분한 것으로 판명되었다. 2015년 US FDA는 이러한 장애물을 극복하기 위해 EAP를 제정했다. EAP는 의료기기를 검토 대기열의 맨 앞에 배치하고 제조업체에게 새로운 규제 접근방식을 촉진하기 위해 US FDA와의 시판 전 협력 기회를 제공했다. BDP는 현재 제조업체가 의료기기 개발과 규제를 형성하는데 있어 US FDA와 협력할 수 있는 추가적인 기회를 제공함으로써 EAP의 교훈을 기반으로 한다(표1).

BDP는 최소한의 임상적으로 의미있는 효과를 반영하기 위해 적응형 연구설계(adaptive study design)와 연구목적 사전 지정(pre-specification of endpoints)과 같은 방법을 포함하여 효율적이고 유연한 임상근거 생성에 중점을 둔다. 안전성 및 효과성에 대한 법적 요건은 변경되지 않지만, US FDA는 이러한 데이터가 판매 승인 시점에 의료기기 성능에 대한 통찰력을 제한할 수 있음을 인식한다. 따라서 US FDA는 제조업체가 요구되는 시판 후 관리 및 연구를 즉시 준수한다는 조건 하에서 시판 전 검토 중에 획기적 의료기기의 위험과 혜택에 대한 더 큰 불확실성을 수용할 것이라고 밝혔다.
초기 BDP 경험(Early BDP experience)
1. 획기적 의료기기 지정 수(Number of breakthrough device designations)
2016년 12월 13일(BDP 개시일)부터 2018년 12월 31일까지 US FDA는 84개 의료기기에 획기성 지정을 부여했다. 2019년 1월 1일부터 2020년 1월 1일까지 지정된 의료기기 수는 거의 3배가 되었고 또 다른 138개 의료기기가 획기성 지정을 받았다(그림1). US FDA는 획기적 의료기기 지정을 위해 모든 제조업체 신청의 70%를 수용했다.

2. 혁신적 의료기기 특징(Breakthrough device characteristics)
2020년 1월 1일 현재, US FDA는 15개 획기성 지정 의료기기를 승인했고, 그중 13개 (86.7%)가 공개되었다. 공개되지 않은 2개 획기적 의료기기는 모두 510(k) 경로를 통해 승인되었다. 공개된 의료기기 가운데 대다수(7/13, 53.8%)는 시판전 허가(pre-market approval, PMA) 경로를 통해 승인된 고위험, 치료용, 이식형 의료기기였다. 2개 치료 의료기기는 510(k) 경로를 통해 승인되었고, 나머지는 PMA(1개, 7.7%) 또는 De Novo(3개, 23.1%) 경로를 통해 승인된 진단 의료기기였다. 공개된 고위험 획기적 의료기기(8개)의 US FDA 검토기간은 중앙값이 181.5일(146~301 일)이었다. 대부분(5/7개, 71.4%) 고위험 치료 획기적 의료기기는 US FDA 승인 이전에 유럽연합에서 이용가능했다(3~15년). US FDA 승인을 받기 전에는 혁신적 진단 의료기기가 해외에 판매되지 않았다.
3. 획기성 지정의 근거(Rationale for breakthrough designation)
획기성 지정을 뒷받침하는 근거는 13개 의료기기 가운데 7개(53.8%)에 대해 공개되었다. 7개 의료기기 가운데 뒷받침하는 기준의 중앙값은 2개(1~3개 기준)였다. US FDA는 이러한 의료기기의 대다수(5개, 71.4%)가 승인된 대체 의료기기에 비해 상당한 이점을 제공한다고 생각했다. US FDA는 승인된 대체 의료기기의 부재(3개, 42.9%), 환자에게 최선의 이익을 위한 이용가능성(3개, 42.9%), 그리고 소수의 의료기기에 대한 획기적 기술(1개, 14.3%)을 포함한 뒷받침하는 기준을 추가로 인용했다.
4. 획기적 의료기기를 뒷받침하는 시판 전 임상 근거(Premarket clinical evidence supporting breakthrough devices)
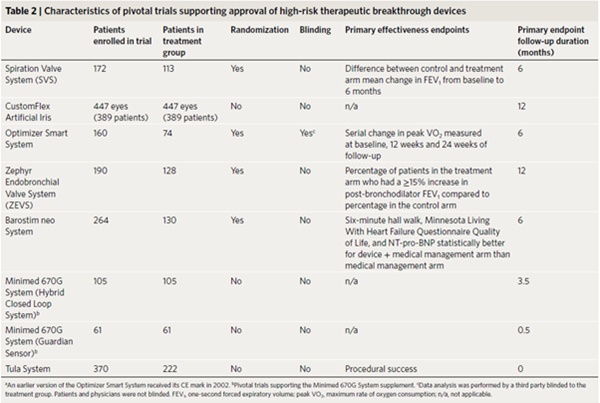
US FDA 문서에는 공개된 획기적 의료기기 13개 가운데 11개(84.6%)에 대한 시판 전 임상근거가 요약되어 있다. 3개(75%) 진단 의료기기는 표준(gold standards)과 관련된 진단 기능을 검증하는 중추적 연구(pivotal studies, 3건)에 의해 뒷받침되었다. 판매 승인을 뒷받침하는 임상연구에 대한 정보가 있는 대부분의 고위험 치료 혁신적 의료기기(6/7개, 83.3%)는 단일 시판 전 중추적 연구에 의해 뒷받침되었다(표2).
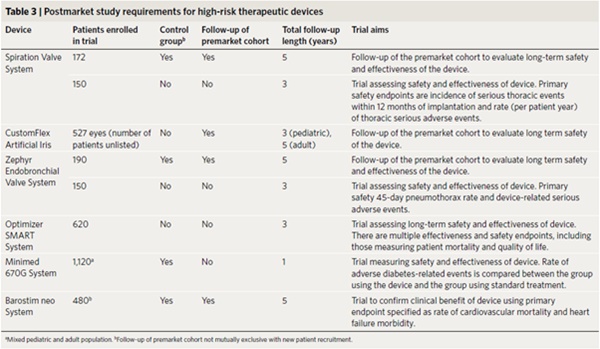
5. 혁신적 치료 의료기기에 대한 시판 후 연구 요건(Post-market study requirements for therapeutic breakthrough devices)
US FDA는 PMA 경로를 통해 승인된 획기적 치료 의료기기 7개 가운데 6개(85.7%)에 대해 시판 후 연구(시판 후 연구 수 1~2건, 중앙값 1)를 요구했다(표3). 시판 후 연구는 승인 당시 의료기기의 위험과 혜택의 불확실성을 반영했다.
|
시사점 |
자료원 : Johnston JL, Dhruva SS, Ross JS, Rathi VK. July 23, 2020. Nature Biotechnology 2000;38:923-938. https://doi.org/10.1038/s41587-020-0636-7
https://www.nature.com/articles/s41587-020-0636-7
* 본 컬럼은 의료기기를 비롯한 헬스케어 분야의 국내외 학회지에 발표된 논문 및 연구보고서 등을 살펴봄으로써 우리나라 의료기기 관련 보건의료정책 마련에 통찰력을 제공하기 위한 목적으로 매주 발표됨